The cardiovascular system is subject to a wide range of diseases and disorders that affect the heart and circulatory system. Acute coronary syndromes (myocardial infarctions, unstable angina) and stroke are highly prevalent causes of morbidity and premature death. Atherosclerosis, angina, peripheral arterial disease, arrhythmia and atrial fibrillation can be debilitating and are potentially life-threatening.
A wide range of different drug classes are used in this branch of medicine. In this module we will summarise the most frequently used classes of drugs that are effective for either prevention or treatment of the most common cardiovascular disorders.
Alpha-adrenoceptor blocking drugs
Alpha-blockers, α-blockers, or α-adrenergic-antagonists are pharmacological agents that act as antagonists of α-adrenoceptors. Drugs can be non-selective, antagonising both α1 and α2 receptor subtypes, or may be selective for one subtype over the other.
α-blockers are most commonly used to treat hypertension, but are also widely used to treat symptoms of BPH (benign prostatic hyperplasia). Raynaud’s disease symptoms are also responsive to α-blocker therapy, although efficacy in this condition has not been clearly established.
Side-effects common to all α-blockers include the risk of hypotensive events (especially following first dose), dizziness, drowsiness, dry mouth and headache. Many side-effects lessen over time.
Non-selective α-blockers
phenoxybenzamine (for control of hypertensive episodes associated with Phaeochromocytoma)
phentolamine (for control of hypertensive episodes associated with Phaeochromocytoma and local anesthetic reversal- not UK approved)
α1-selective blockers
alfuzosin (symptomatic relief in benign prostatic hyperplasia, BPH)
prazosin (BPH, hypertension)
doxazosin (BPH, hypertension)
terazosin (BPH, hypertension)
silodosin (BPH)
tamsulosin (BPH)
indoramin (BPH)
α2-selective blockers
mirtazapine (treatment of episodes of major depression)
Angiotensin-converting enzyme (ACE) inhibitors
Angiotensin II (ATII) is a highly potent endogenous vasoconstrictor. It is formed from angiotensin I in the blood by the angiotensin converting enzyme (ACE). The renin–angiotensin-aldosterone system (RAAS) modulating actions of ATII are mediated by the angiotensin receptors AT1 and AT2.
ACE inhibitors partially prevent the conversion of ATI to ATII. As a result, blood vessels dilate, and blood pressure is reduced. Reducing blood pressure makes it easier for the heart to pump blood and can improve the function of a failing heart. An additional clinical intervention delivered by ACE inhibitors is slowing progression of kidney disease due to high blood pressure or diabetes.
ACE inhibitors are the first-line choice in hypertension treatment, especially in those under 55 years old. Using ACE inhibitors to treat heart failure is not so age dependent, and the level of benefit achieved seems to increase with age. Angiotensin receptor antagonist type drugs are often used in patients intolerant to ACE inhibitors, and prove useful as they inhibit the activity of residual ATII present in the blood due to incomplete ACE inhibition. The combination therapy of angiotensin receptor antagonists with ACE inhibitors may be superior to either agent alone.
ACE inhibitors are administered as prodrugs or prodrug conjugates, which are metabolised in the liver to the active compound. ACE inhibitor prodrugs have the generic name stem suffix ‘-pril’, and the active compound has the stem ‘-prilat’.
Captopril was the first ACE inhibitor to be approved, but has since been superseded by alternatives with improved duration of action and fewer adverse effects. There is a wide choice of ACE inhibitors available to the prescriber, but all have similar antihypertensive activity at equivalent doses. The main difference between the different drugs is their duration of action. Some of those available are listed below, along with equivalent dose information.
ACE inhibitors dosages for hypertension
| Name | Eq. daily dose | Start dose | Usual dose | Max. dose |
|---|---|---|---|---|
| benazepril | 10mg | 10mg | 20-40mg | 80mg |
| captopril | 50mg (25mg bid) | 12.5-25mg bid-tid | 25–50mg bid-tid | 450mg/day |
| enalapril | 5mg | 5mg | 10-40mg | 40mg |
| fosinopril | 10mg | 10mg | 20-40mg | 80mg |
| lisinopril | 10mg | 10mg | 10-40mg | 80mg |
| moexipril | 7.5mg | 7.5mg | 7.5-30mg | 30mg |
| perindopril | 4mg | 4mg | 4-8mg | 16mg |
| ramipril | 2.5mg | 2.5mg | 2.5-20mg | 20mg |
| trandolapril | 2mg | 1mg | 2-4mg | 8mg |
| quinapril | 10mg | 10mg | 20-80mg | 80mg |
| tid = three times a day | bid = two times a day | |||
All ACE inhibitors are contra-indicated in patients with previous ACE inhibitor-induced angiodema, or hypersensitivity to the drug. Prescribers should be cautious of prescribing ACE inhibitors to patients with renal impairment, aortic valve stenosis or cardiac outflow obstruction, hypovolemia, dehydration or those on hemodialysis (using high-flux polyacrylonitrile membranes). Administration of ACE inhibitors to women who are likely to become pregnant should be avoided, as should use during pregnancy due to risk of drug-induced birth defects. A mnemonic to aid recollection of ACE inhibitor contra-indications is PARK: Pregnancy, Allergy/Angiodema, Renal failure, K– hyperkalemia (potassium >5.5).
Side effects include a dry, irritating cough and more rarely renal stenosis or allergic reaction.
RESOURCES
 | Renin Angiotensin Aldosterone System (RAAS) – Short and sweet!
| Renin Angiotensin Aldosterone System (RAAS) – Short and sweet!Angiotensin receptor antagonists
Angiotensin receptor antagonists are used to treat hypertension, diabetic nephropathy and congestive heart failure. As a group these drugs are termed ‘sartans’, and this forms the stem of their non-proprietary names.
Clinical angiotensin receptor antagonists inhibit angiotensin II-induced activation of the AT1 receptor, thereby modulating the renin–angiotensin system. This action leads to an inhibition of the effects mediated by angiotensin II, principally vasoconstriction and aldosterone and vasopressin secretion, which manifest clinically as decreased blood pressure and vascular resistance.
The efficacy of each sartan depends on its pressor inhibition (inhibition of the blood pressure-raising effect of angiotensin II), its AT1 receptor affinity and its biological half-life.
Angiotensin receptor antagonists can be prescribed as single agents or in fixed-dose combinations with other antihypertensive agents, in particular with the thiazine diuretic hydrochlorothiazide.
All angiotensin receptor antagonists are contra-indicated with the direct renin inhibitor aliskiren, in patients with diabetes mellitus or who have kidney disease with an eGFR <60. All of these drugs should be avoided in patients with severe hepatic impairment and in pregnancy unless essential as they may adversely affect fetal and neonatal blood pressure control and renal function. Use with caution, in patients with renal impairment, starting with a low dose, and titrating according to response.
Single agent drugs:
Telmisartan: indicated for hypertension (20–40 mg once daily, titrating to 80mg if necessary), prevention of cardiovascular events in patients with established atherosclerotic cardiovascular disease, or diabetic organ damage (80mg once daily). Common side-effects include arthralgia, back pain, chest pain, eczema, gastro-intestinal disturbances, influenza-like symptoms, leg cramps, myalgia, pharyngitis, sinusitis and urinary-tract infection.
Olmesartan/Olmesartan medoxomil: Indicated for hypertension, starting at a daily dose of 10mg, increasing to 20mg if necessary. Maximum dose is 40mg/day. Contra-indicated in patients with biliary obstruction. Side-effects include arthritis, chest pain, cough, fatigue, gastro-intestinal disturbances, haematuria, hypertriglyceridaemia, hyperuricaemia, influenza-like symptoms, musculoskeletal pain, peripheral oedema, pharyngitis, rhinitis, and urinary-tract infection.
Valsartan: Indicated for hypertension, heart failure when ACE inhibitors cannot be used, or in conjunction with an ACE inhibitor when a beta-blocker cannot be used and myocardial infarction with left ventricular failure or left ventricular systolic dysfunction. Additional specific contra-indications arebiliary cirrhosis and cholestasis. Common side-effect is renal impairment.
Losartan: potassium: Indicated for hypertension, heart failure when ACE inhibitors cannot be used and diabetic nephropathy in type 2 diabetes mellitus. Additional specific contra-indication is for severe heart failure. Vertigo is a common side-effect. The manufacturer advises avoidance in patients with severe hepatic impairment
Irbesartan: Indicated in patients with hypertension, haemodialysis patients with hypertension and patients with type 2 diabetes renal disease. Common side-effects include fatigue, musculoskeletal pain, nausea and vomiting.
Azilsartan/azilsartan medoxomil: Indicated for hypertension and hypertension with intravascular volume depletion. Diarrhoea and raised creatine kinase levels are common side-effects. The manufacturer advises avoidance in patients with severe hepatic impairment, and low dose and close monitoring in patients with mild to moderate hepatic impairment.
Eprosartan: Indicated for hypertension, with an adult dose of 600mg once daily. Common side-effects include headache, nausea, rhinitis, diarrhoea, malaise and vomiting.
Candesartan/candesartan cilexetil: Indicated for hypertension (usual dose 8 mg once daily), hypertension with intravascular volume depletion (usual dose 8 mg once daily), heart failure with impaired left ventricular systolic function when ACE inhibitors are not tolerated and heart failure with impaired left ventricular systolic function in conjunction with an ACE inhibitor, under expert supervision (dosage starting at 4 mg once daily, increasing fortnightly to a target dose of 32 mg once daily or to the maximum tolerated dose). Contra-indicated in patients with cholestasis. In common with other sartans, common side-effects are headache and vertigo. Avoid prescribing in patients with severe hepatic impairment and use caution in renal impairment (eGFR less than 15).
Fixed-dose combination medicines:
Losartan with hydrochlorothiazide
Irbesartan with hydrochlorothiazide
Amlodipine with valsartan
Valsartan with hydrochlorothiazide
Olmesartan with amlodipine (a calcium-channel blocker)
Telmisartan with hydrochlorothiazide
Olmesartan with hydrochlorothiazide
Olmesartan with amlodipine and hydrochlorothiazide
This review article by Aulakh et al. (2007) discusses various angiotensin receptor antagonists.
Anti-arrhythmic drugs
Antiarrhythmic drugs are a group of pharmaceuticals that are used to suppress abnormal rhythms of the heart (cardiac arrhythmias), such as atrial fibrillation, atrial flutter, ventricular tachycardia, and ventricular fibrillation.
Class I antiarrhythmics interfere with sodium channel function, and are subdivided by the effect they have on the action potential (AP)- see figure below
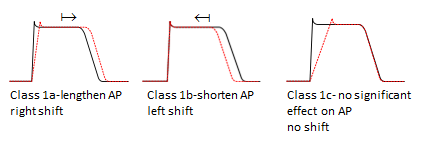
Class 1a antiarrhythmics
e.g. disopyramide (used for the prevention and treatment of ventricular and supraventricular arrhythmias and maintenance of sinus rhythm after cardioversion)
Class 1b antiarrhythmics
e.g. lidocaine (as well as being a local anaesthetic, lidocaine can be used to treat ventricular arrhythmias, and as an alternative for cardiopulmonary resuscitation if amiodarone is not available)
Class Ic antiarrhythmics
e.g. flecainide (used to treat various arrhythmias: AV nodal reciprocating tachycardia, arrhythmias associated with Wolff-Parkinson-White syndrome and supraventricular arrhythmias) and propafenone (mainly used under specialist supervision in hospital to treat supraventricular arrhythmias and paroxysmal supraventricular tachyarrhythmias which include paroxysmal atrial flutter or fibrillation and paroxysmal re-entrant tachycardias involving the AV node or accessory pathway, where standard therapy ineffective or contra-indicated).
Class II antiarrhythmics are conventional beta blockers- See the beta-adrenoceptor blocking drugs topic above this one
Class III antiarrhythmics (primarily affect potassium (K+) efflux, prolonging repolarization)
e.g. dronedarone (used to treat life-threatening atrial flutter and fibrilation, and to maintain sinus rhythm after cardioversion) and amiodarone (used to treat angina and arrhythmias). Note that amiodarone should not be given alongside sofosbuvir with daclatasvir, sofosbuvir and ledipasvir, and simeprevir with sofosbuvir due to a risk of severe bradycardia and heart block.
Class IV antiarrhythmics (are slow calcium channel blockers, decrease AV node conduction and shorten AP plateau)
Class V (other) antiarrhythmics
e.g. digoxin (decreases speed of conduction through AV node, used to treat congestive heart failure and atrial fibrillation), adenosine (used intravenously for terminating supraventricular tachycardias), magnesium sulfate (used to treat torsades de pointes)
In relation to the classification above, and management of atrial fibrillation, class I and III antiarrhythmics are used in rhythm control as medical cardioversion agents while class II and IV antiarrhythmics are used as rate control agents.
Anticoagulant drugs
Anticoagulants are used to prevent the formation or growth of fibrin/erythrocyte thrombi in the venous circulation. They are not useful for treating arterial thrombi which are mainly composed of platelet aggregates.
Anticoagulants can be administered parenterally or orally.
Parenteral anticoagulants:
Heparin (unfractionated or ‘standard’ heparin) acts rapidly, but also has a short duration of action. This characteristic allows heparin infusion to be used in patients at high risk of bleeding as the anticoagulation effect ends rapidly upon stopping infusion.
Low molecular weight heparins are as effective as unfractionated heparin, but have a lower risk of causing heparin-induced thrombocytopenia. They are also more convenient to use due to their longer duration of action than unfractionated heparin, and are therefore the preferred option for prevention of venous thromboembolism, for example in the treatment of deep-vein thrombosis (DVT), pulmonary embolism (PE), myocardial infarction (MI), unstable coronary artery disease and for the prevention of clotting in extracorporeal circulatory procedures. Example drugs are dalteparin sodium, enoxaparin sodium, and tinzaparin sodium
Heparinoids, such as danaparoid sodium are mixtures of heparin derivatives termed glycosaminoglycans, such as heparan, dermatan, and chondroitin sulphates. Used to reduce the risk of surgical patients of developing DVT.
Argatroban is an oral anticoagulant that acts as a thrombin inhibitor. It is indicated for prophylaxis or treatment of thrombosis in patients with, or at risk of, heparin-induced thrombocytopenia (HIT).
Hirudins, such as desirudin, lepirudin and bivalirudin are derivatives of the salivary anticoagulant found in leeches. These compounds are direct thrombin inhibitors. Bivalirudin is indicated for unstable angina or non-ST-segment elevation MI in patients planned for urgent or early intervention, and as an anticoagulant for patients undergoing percutaneous coronary intervention.
Epoprostenol (prostacyclin PGI2) can be used to prevent platelet aggregation during renal dialysis if heparins are unsuitable or contraindicated. Epoprostenol has a very short half-life so must be administerd by continuous intravenous infusion. Can also be used in conjunction to oral anticoagulation to treat primary pulmonary hypertension resistant to other treatment, under specialist care.
Fondaparinux inhibits activated coagulation factor X and is indicated for prevention of venous thrombus formation in various at-risk patient groups
Oral anticoagulants:
Coumarins (warfarin and acenocoumarol) and phenindione antagonise the effect of vitamin K, but are slow in achieving full anticoagulatory effect (48-72 hours). From this group of drugs, warfarin is the drug of choice. Warfarin is used as prophylaxis of venous thromboembolism in at-risk patients. Keeling D et al. (2011, PMID: 21671894) provide guidelines on using warfarin anticoagulation.
Dabigatran, rivaroxaban and apixaban are oral anticoagulants which are alternatives to warfarin. Rivaroxaban and apixaban inhibit platelet activation and fibrin clot formation via direct, selective and reversible inhibition of Factor Xa. Dabigatran etexilate is a prodrug that is converted to the active dabigatran in vivo. Dabigatran is a specific reversible direct thrombin inhibitor. Regular laboratory testing is not required for either of these drugs. Additionally, there are fewer drug and food interactions with these oral anticoagulants. These represent advantages to the use of these drugs over warfarin.
Antiplatelet drugs
Antiplatelet drugs are designed to decrease platelet aggregation to inhibit thrombus formation in the arterial circulation. Used to treat patients with cardiac and cerebrovascular conditions.
They are classified according to their mechanism of action:
- Irreversible cyclooxygenase inhibitors– aspirin, used for primary prevention of (cardio)vascular disease in older at risk patients, acute indications and secondary prevention
- Adenosine diphosphate (ADP) receptor inhibitors– clopidogrel, used for acute indications and secondary prevention; prasugrel (plus aspirin), used for the prevention of atherothrombotic events in patients with acute coronary syndrome undergoing percutaneous coronary intervention (PCI); ticagrelor (plus aspirin), used for the prevention of atherothrombotic events in patients with acute coronary syndrome; cangrelor (plus aspirin), is to be used under expert supervision only, in patients undergoing PCI who have not previously received clopidogrel, prasugrel or ticagrelor and for whom these drugs are not suitable.
- Phosphodiesterase inhibitors– cilostazol is indicated for the improvement of the maximal and pain-free walking distances in patients with intermittent claudication. Dose reduction is advised in patients receiving medicines which inhibit CYP3A4 and CYP2C19. Cilostazol is contraindicated in patients with moderate or severe hepatic impairment, and those with creatinine clearance of ≤ 25 ml/min.
- Protease-activated receptor 1 (PAR1) antagonists– vorapaxar is not approved in the UK, but is used elsewhere to reduce the risk of thrombotic cardiovascular events in patients with a history of myocardial infarction (MI) or with peripheral arterial disease (PAD). Contraindicated in patients with a history of stroke, transient ischemic attack (TIA), or intracranial hemorrhage (ICH), or with active bleeding. Metabolism is mainly hepatic, so use is not recommended in patients with severe liver impairment.
- Glycoprotein IIB/IIIA inhibitors block the binding of fibrinogen to receptors on platelets- abciximab, used as an adjunct to unfractionated heparin and aspirin for the prevention of ischaemic complications in high-risk patients undergoing percutaneous transluminal coronary intervention, as a once only treatment; eptifibatide (plus unfractionated heparin and aspirin) and tirofiban (plus unfractionated heparin, aspirin, and clopidogrel) are used to prevent early myocardial infarction in patients with unstable angina or non-ST-segment-elevation myocardial infarction, and the tirofiban combination therapy can be used in non-ST-segment-elevation myocardial infarction patients undergoing PCI.
- Adenosine reuptake inhibitors– dipyridamole, used as an adjunct to oral anticoagulation for prophylaxis of thromboembolism associated with prosthetic heart valves and for secondary prevention of ischaemic stroke and transient ischaemic attacks.
- Thromboxane receptor antagonists– none are approved but see Davı et al. (2012) for rationale behind their development
- Thromboxane synthase inhibitors– none are approved but see Davı et al. (2012) for rationale behind their development
RESOURCES
Thromboxane Receptors Antagonists and_or Synthase Inhibitors_Davietal_2012_.pdf
This comprehensive review discusses the pathophysiological rationale for the expected superiority of TP receptor antagonists over aspirin as anti-thrombotic agents, as well as providing an overview of the development of thromboxane receptor antagonists, and their failure to reach approval.
Beta-adrenoceptor blocking drugs
Beta blockers, also known as beta-adrenoceptor blocking agents, are pharmaceutical agents that act as competitive antagonists of the β-adrenoceptors, which are activated by the endogenous catecholamines epinephrine (adrenaline) and norepinephrine (noradrenaline), of the sympathetic nervous system. Beta blocking drugs used to treat conditions such as angina, heart failure, cardiac arrhythmias, edema and high blood pressure and can also be used in the management of glaucoma and anxiety.
Side-effects are generally not serious, and lessen as the body adjusts to treatment. Hypotensive effects (especially following first dose), tiredness, dizziness and cold hands and feet are quite common side-effects. In men, beta blocker use can cause erectile dysfunction.
Beta blockers are contra-indicated in patients suffering from asthma, cardiogenic shock, hypotension, marked bradycardia, metabolic acidosis, phaeochromocytoma (apart from specific use with alpha-blockers), Prinzmetal’s angina, second-and third-degree AV block, severe peripheral arterial disease, sick sinus syndrome or uncontrolled heart failure. Additionally, beta blockers, including those considered to be cardioselective, should be avoided where possible in patients with a history of asthma, bronchospasm or a history of obstructive airways disease. If there is no alternative, cardioselective beta blockers can be prescribed to patients under specialist care, with particular attention paid to the induction of bronchospasm. There is a risk of precipitating heart failure when beta blockers and the calcium channel blocker (antiarrhythmic), verapamil, are used together in established ischaemic heart disease.
LIke α-blockers, beta blockers can be selective or non-selective for β-adrenoceptor subtypes. β1-selective drugs are considered to be cardioselective as the heart and kidneys are the main sites of β1-adrenoceptor expression. β2-adrenoceptors are expressed in the lungs, gastrointestinal tract, liver, uterus, vascular smooth muscle, and skeletal muscle, and β3-adrenoceptors are found in fat cells.
Non-selective beta blockers
timolol (hypertension, angina, migraine prophylaxis, prophylaxis after myocardial infarction, glaucoma- reduces elevated intraocular pressure)
pindolol (hypertension, angina)
propranolol (hypertension, angina, anxiety, essential tremor, migraine prophylaxis, arrhythmias, prophylaxis after myocardial infarction, management of the symptoms of thyrotoxicosis)
levobunolol (glaucoma- reduces elevated intraocular pressure)
carteolol (hypertension, elevated intraocular pressure)
sotalol (cardiac arrythymias)
oxprenolol (angina, hypertension and cardiac arrythymias)
nadolol (angina, hypertension, migraine pain and tremor)
Selective beta blockers (β1-selective, cardioselective)
atenolol (hypertension, angina, arrhythmias)
esmolol (tachycardia and hypertension in peri-operative period, short-term treatment of supraventricular arrhythmias)
metoprolol (hypertension, angina, arrhythmias, early intervention within 12 hours of infarction, migraine prophylaxis)
celiprolol (mild-moderate hypertension)
co-tenidone (hypertension), co-tenidone is a 4:1 mixture of atenolol and the carbonic anhydrase inhibitor, chlortalidone
bisoprolol (hypertension, angina)
acebutolol (hypertension, angina, arrhythmias)
nebivolol (essential hypertension, hypertension in patients with renal impairment, adjunct in stable mild to moderate heart failure)
betaxolol (hypertension, primary open-angle glaucoma) note that β1 selectivity is not absolute, and some inhibitory effects on β2-adrenoceptors is exhibited at higher doses.
This webpage provides information around the history and development of beta blocker drugs, their pharmacology, mechanism of action, pharmacokinetics and adverse effects. Produced by Pharmafactz. The website also includes short quizzes on various pharmacological topics and medication areas.
Calcium channel blocking drugs
Calcium-channel blockers (CCBs) are prescribed to treat hypertension. These drugs interfere with the inward movement of calcium ions through the slow channels on the membranes of myocardial cells, AV node cells and vascular smooth muscle cells. The overall effect is to reduce myocardial contractility, formation and propagation of electrical impulses within the heart, and vascular tone.
There is a wide variety of CCB medications available to prescribers, including single agent medicines and fixed-dose combination drugs.
Common side effects include dizziness, flushing, headache, oedema and palpitation.
For all CCBs, prescribers should be alert to the possibility of overdose (CCB poisoning), with symptoms including nausea, vomiting, dizziness, agitation, confusion, coma in severe poisoning, metabolic acidosis and hyperglycaemia. It is advised that treatment cessation should be gradual, as sudden withdrawal may be associated with an exacerbation of myocardial ischaemia.
All CCBs should be avoided in pregnancy, unless the risk of uncontrolled maternal hypertension outweighs the risk to the fetus. Due to hepatic metabolism, dose in patients with liver impairment may need to be reduced.
Single agent drugs:
Amlodipine: an oral dihydropyridine type CCB prescribed for angina and hypertension. Contra-indicated in patients suffering cardiogenic shock, significant aortic stenosis or unstable angina.
Felodipine: an oral dihydropyridine type CCB prescribed for angina and hypertension. Use is contra-indicated in patients with cardiac outflow obstruction, significant cardiac valvular obstruction (e.g. aortic stenosis), uncontrolled heart failure, unstable angina or within 1 month of myocardial infarction.
Isradipine: an oral CCB prescribed for hypertension. Use is contra-indicated in patients with acute porphyrias, cardiogenic shock, during or within 1 month of myocardial infarction or unstable angina.
Lacidipine: an oral dihydropyridine type CCB prescribed for hypertension. Contra-indicated in the same patients as isradipine.
Lercanidipine: an oral dihydropyridine type CCB prescribed for mild to moderate hypertension. Contra-indicated in the same patients as isradipine.
Nicardipine: an oral dihydropyridine type CCB prescribed in immediate-release tablet form for mild to moderate hypertension and prophylaxis of angina. May be delivered intravenously by specialists in patients with life-threatening hypertension in several scenarios (post-operatively, in patients with hepatic or renal impairment or with acute life-threatening hypertension in pregnancy). Use is contra-indicated in patients with acute porphyrias, cardiogenic shock, significant or advanced aortic stenosis or unstable or acute attacks of angina. Oral administration is to be avoided within 1 month of myocardial infarction (MI), intravenous administration to be avoided within 8 days of MI.
Nifedipine: an oral dihydropyridine type CCB prescribed to treat hypertension and Raynaud’s syndrome. Not generally recommended for angina prophylaxis. May be used off-label to postpone premature labour. Contra-indicated in the same patients as isradipine.
Nisoldipine: (not available in the UK). An oral dihydropyridine type CCB indicated for hypertension that can be used alone or in combination with other antihypertensive agents. Avoid coadministration with CYP3A4 modulating drugs.
Nimodipine: an oral or intravenously administered dihydropyridine type CCB indicated for the treatment or prevention of ischaemic neurological defects following aneurysmal subarachnoid haemorrhage. Contra-indicated in patients with acute porphyrias or unstable angina, or within 1 month of myocardial infarction.
Diltiazem: an oral CCB indicated for angina and mild to moderate hypertension in extended-release formulations. Contra-indicated in patients suffering acute porphyrias, left ventricular failure with pulmonary congestion, second- or third-degree AV block (unless pacemaker fitted), severe bradycardia and sick sinus syndrome.
Verapamil: indicated for the treatment of supraventricular arrhythmias (by mouth or slow intravenous injection), paroxysmal tachyarrhythmias (slow intravenous injection), angina (by mouth), hypertension (immediate-release oral medicines) and prophylaxis of cluster headache (initiated under specialist supervision, using immediate-release oral medicines). Oral verapamil may also be used as prophylaxis after myocardial infarction in patients for whom beta-blockers are not appropriate. Use is contra-indicated in patients with acute porphyrias, atrial flutter or fibrillation associated with accessory conducting pathways (e.g. Wolff-Parkinson-White-syndrome), bradycardia, cardiogenic shock, a history of heart failure (even if controlled by therapy), a history of significantly impaired left ventricular function (even if controlled by therapy), hypotension, second- and third-degree AV block, sick sinus syndrome or sino-atrial block. Constipation is a common side-effect.
Fixed-dose combination drugs:
Nifedipine with atenolol: CCB in combination with a beta-adrenoceptor antagonist (beta-blocker) prescribed for hypertension and angina. Only indicated when calcium-channel blocker or beta-blocker alone proves inadequate
Amlodipine with valsartan: CCB in combination with an angiotensin AT1 receptor antagonist prescribed for hypertension in patients who are already stabilised on the individual drugs at the same doses.
Amlodipine with olmesartan: CCB in combination with an angiotensin AT1 receptor antagonist prescribed for hypertension in patients who are already stabilised on the individual drugs at the same doses.
Amlodipine with olmesartan and hydrochlorothiazide: CCB in combination with an angiotensin AT1 receptor antagonist and a thiazide diuretic prescribed for patients who are already stabilised on the individual drugs at the same doses, or in whom hypertension is not adequately controlled by olmesartan and amlodipine.
Felodipine with ramipril: CCB in combination with an angiotensin-converting enzyme (ACE) inhibitor pro-drug prescribed for patients who are already stabilised on the individual drugs at the same doses.
RESOURCES
The website run by bloodpressureuk.org contains both patient and professional oriented materials. This specific page provides links to guidelines produced by professional societies and associations, and national/international agencies, including NICE (The National Institute for Health and Care Excellence) and the World Health Organization.
Diuretics
Diuretics are particularly useful in treating the water and salt retention caused by heart failure.
Common side effects like muscle cramps arise from the loss of too much sodium, potassium or magnesium. Prescribers should be aware of the risk of diuretic-induced hypokalaemia.
There are three main types of diuretic, each of which works by affecting a different part of the kidneys:
Loop diuretics– prescribed for fluid retention (oedema), particularly in the lungs. These drugs are very fast acting, but of short duration and induce dramatic water loss.
e.g. furosemide, bumetanide and occasionally torasemide.
Furosemide can be prescribed with potassium chloride to maintain potassium levels.
Thiazide diuretics– fast acting but of longer duration than the loop diuretics, with less dramatic water loss. These are commonly used at low dose in the management of hypertension.
e.g. bendroflumethiazide, hydrochlorothiazide and indapamide
Potassium-sparing diuretics– these are a weaker type of diuretic which increase water loss, but prevent loss of too much potassium. Potassium-sparing diuretics are usually prescribed to treat water retention due to heart failure
e.g. spironolactone and eplerenone (aldosterone antagonists), triamterene and amiloride (act on epithelial sodium channels, ENaCs)
Potassium-sparing diuretics are not usually necessary in the routine treatment of hypertension, unless hypokalaemia develops.
Other diuretics
Osmotic diuretics such as mannitol, given by intravenous (IV) infusion, can be used to treat cerebral oedema and raised intraocular pressure (e.g. in glaucoma patients).
Carbonic anhydrase inhibitors such as acetazolamide (administered orally or IV) and dorzolamide and brinzolamide (topically applied) are diuretics used to treat glaucoma.
Drugs from these different families can be prescribed in fixed-dose combinations, which can be used when compliance using separate medications is an issue. Examples include furosemide with triamterene or spironolactone, co-triamterzide (triamterene plus hydrochlorothiazide), co-amilofruse (amiloride plus furosemide) and co-amilozide (amiloride plus hydrochlorothiazide). Dorzolamide and brinzolamide can be prescribed in fixed-dose combination with the beta blocker timolol, for the management of raised intraocular pressure and glaucoma, in patients for whom single agent beta blocker therapy is inadequate.
RESOURCES
A 16 minute video re-capping the structure/functions of the kidney, followed by an overview of the action of different types of diuretics. Produced by Armando Hasudungan (http://armandoh.org/).
A 9 minute video reviewing the structure of the nephron and the location and action of diuretics.
This is a slide set (13 slides) covering osmotic diuretics and carbonic anhydrase inhibitors. It also discusses aldosterone and vasopression activity in the kidney, as well as the mechanism of action of SGLT2 inhibitors. The final slides discuss gout and the mechanisms employed by current anti-gout medications. It is an updated version for the 2017-18 academic year. Provided by Prof. JA Peters, University of Dundee School of Medicine.
This a is a slide set (22 slides) covering some of the drugs acting in the kidney. The presentation begins with a general overview, then outlines the causes of odema, and diseases with fluid retention which respond to diuretics (slides 5 & 6). Following a slide illustrating the main sites of diuretic action in the nephron, loop and thiazide diuretics are discussed along with the mechanism of potassium loss from the kidney. The final slides describe the action and uses of potassium sparing diuretics. This is an updated version of the lecture series for the 2017-18 academic year. Provided by Prof. JA Peters, University of Dundee School of Medicine.
Fibrinolytic drugs
Fibrinolytic and thrombolytic drugs are used to break down blood clots (thrombi) to limit occlusion of blood vessels. Fibrinolytic drugs act as thrombolytics by activating plasminogen to form plasmin, which breaks down fibrin in the thrombus. These types of drugs are used to treat myocardial infarction, thromboembolic strokes, deep vein thrombosis and pulmonary embolism to limit damage to the perfused tissue, and ultimately to prevent death from these conditions. They are most effective when administered as soon as diagnosis determines their use to be beneficial. Often used synergistically with anticoagulants such as heparin. Fibrinolytics are contraindicated in hemorrhagic stroke, and other indications where bleeding is a risk factor (including cavitary pulmonary disease, acute pancreatitis, aneurysm, aortic dissection, bleeding diatheses, coagulation defects, heavy vaginal bleeding, history of cerebrovascular disease or recent haemorrhage, oesophageal varices, peptic ulceration, recent trauma and severe hypertension).
Thrombolytic drugs include:
- Recombinant tissue plasminogen activator proteins (r-tPAs) such as alteplase, reteplase and tenecteplase. All three are indicated for acute myocardial infarction (AMI), with alteplase also being indicated for pulmonary embolism (PE), stroke, and thrombolytic treatment of occluded central venous access devices.
- anistreplase (anisoylated plasminogen streptokinase activator complex (APSAC)- an acylated complex of purified human plasminogen and bacterial streptokinase. Upon hydrolysis of the acyl group, the activator complex converts plasminogen to plasmin, which breaks down fibrin.
- streptokinase– a bacterial enzyme which activates plasminogen to produce a thrombolytic effect. Indicated for AMI, deep vein thrombosis (DVT), PE, acute arterial thromboembolism and central retinal venous or arterial thrombosis.
- urokinase (urokinase-type plasminogen activator (uPA))- a serine protease that activates plasminogen to break down thrombi. Indicated for DVT, PE, occlusive peripheral arterial disease, occluded central venous catheters and occluded arteriovenous haemodialysis shunts.
The summary table below is reproduced from Pharmafactz.com.
Summary of fibrinolytics used in the clinic.
| FIBRINOLYTIC DRUGS | |||
|---|---|---|---|
| Fibrinolytic drugs enhance the normal process of thrombus degradation, via activation of plasminogen, an enzyme crucial to clot breakdown. | |||
| Mechanistically they are in effect recombinant versions of tissue plasminogen activator (t-PA), the enzyme that converts plasminogen to plasmin- plasmin being largely responsible for thrombus degradation. | |||
| Alteplase is a genetically modified, recombinant version of t-PA which binds to fibrinogen and fibrin. | Tenecteplase is a recombinant, modified t-PA with improved fibrin specificity, and longer duration of action than t-PA. | Reteplase is a recombinant t-PA which binds less to fibrinogen and fibrin, but has a longer duration of action than the endogenous enzyme. | Streptokinase is derived from haemolytic streptococci. It is inactive until it binds to circulating plasminogen. |
|
Side effects Haemorrhage- particularly intracerebral haemorrhage occurs in ~1% of patients. Hypotension- dose related, and more common with streptokinase. Allergic reactions to streptokinase. |
All fibrinolytic drugs are administered IV or intra-arterially. Half-life of streptokinase (1h) is longer than alteplase or reteplase (0.5h), but similar to tenecteplase. |
||
Lipid-lowering drugs
There are several classes of established lipid-lowering, or antihyperlipidemic drugs available to the prescriber.
The choice of which agent to use depends greatly on the patient’s cholesterol profile, cardiovascular, liver and kidney function.
Statins are designed to lower LDL, the ‘bad cholesterol’ most strongly linked to vascular disease. Notable side-effects include myopathy and rhabdomyolysis. Some evidence suggests that statins should not be used in patients older than 75 who have no history of heart disease or stroke. Statins inhibit the enzyme hydroxymethylglutaryl-CoA reductase (HMGCR). Examples of approved statins include atorvastatin, cerivastatin, fluvastatin, lovastatin, pravastatin, rosuvastatin and simvastatin.
Fibrates are prescribed to reduce hypercholesterolemia in patients intolerant of or unsuitable for statin therapy. Fibrates increase HDL (‘good cholesterol’) and lower triglyceride levels. The fibrate-induced reduction in insulin resistance is useful when the dyslipidemia is associated with other indicators of the metabolic syndrome (e.g. hypertension and type 2 diabetes mellitus). Fibrate therapy is unsuitable for patients with low HDL levels, and treatment should be withdrawn if HDL-C levels are severly depressed soon after initiation. Fibrates are recommended as first line therapy only in patients with very high triglyceride levels. Fibrates activate peroxisome proliferator-activated receptors (PPARs), especially PPARα. They are metabolised by cytochrome P450 3A4 (CYP3A4). Examples of approved fibrates include bezafibrate, ciprofibrate, clofibrate, fenofibrate and gemfibrozil.
Niacin (nicotinic acid or vitamin B3) is used as a vitamin supplement. It lowers both cholesterol and triglyceride concentrations by inhibiting synthesis; it also increases HDL cholesterol. Used as an adjunct to statin therapy, or alone if statins are not tolerated. Not to be used if the patient has a history of peptic ulcer disease or arterial bleeding. The nicotinic acid derivative acipimox can be used in similar situations. Inositol nicotinate can be used for peripheral vascular disease, but not in patients in the acute phase of a cerebrovascular accident or who have suffered recent myocardial infarction. NICE does not recommend the use of inositol nicotinate to treat intermittent claudication in patients with peripheral arterial disease.
Bile acid sequestrants are synthetic polymeric resins which prevent reabsorption of bile constituents from the gut, thereby acting a hypolipidemic agents. Also used for other purposes such as the treatment of chronic diarrhea due to bile acid malabsorption and for the prevention of pruritus in patients with chronic liver disease. As hypolipidemic agents these are less effective than statins. Not to be used when blood triglyceride levels are elevated. Examples include cholestyramine, colestipol and colesevelam.
Ezetimibe inhibits the intestinal cholesterol uptake protein Niemann-Pick C1-like protein 1, a critical mediator of intestinal cholesterol absorption. Has only a modest effect as a mono-therapy, but can be used adjunctively with dietary measures with or without statins to treat primary hypercholesterolaemia and homozygous familial hypercholesterolaemia. Avid use in patients with moderate and severe hepatic impairment.
Lomitapide inhibits the microsomal triglyceride transfer protein (MTTP) resulting in reduced lipoprotein secretion and circulating concentrations of lipoprotein-borne lipids such as cholesterol and triglycerides. Only to be used under expert supervision in patients with homozygous familial hypercholesterolaemia, as an adjunct to dietary measures and other lipid-regulating drugs. Since lomitapide can interfere with the absorption of fat-soluble nutrients, vitamin E and fatty acids supplements are essential.
Phytosterols (plant sterols and stanols) are naturally occurring steroid-like compounds similar to cholesterol. Stanols are saturated sterols, having no double bonds in the sterol ring structure. Despite being effective in lowering LDL cholesterol, the benefits of phytosterol-enriched foods and dietary supplements in cardiovascular disease and overall mortality are yet to be proven [Genser et al. (2012)]. The most commonly occurring phytosterols in the human diet are β-sitosterol, campesterol and stigmasterol; the most common stanols are sitostanol and campestanol.
Orlistat inhibits pancreatic lipase, thereby reducing absorption of fats from the diet. Used as an adjunct to a reduced-calorie diet as an anti-obesity therapy, in obese patients suffering additional risk factors such as type 2 diabetes, hypertension, or hypercholesterolaemia.
RESOURCES
This a is a slide set (42 slides) covering clinically used drugs for lipid lowering. This is an updated version of the lecture series for the 2021-2022 academic year. It is suitable for intermediate level learners.
Contributed by Dr. Zoltan Varga, Semmelweis University (Hungary)
Nitrates
Nitrate medicines include glyceryl trinitrate (GTN), isosorbide dinitrate and isosorbide mononitrate, prescribed under a number of trade names. Nitrates cause vasodilation by activating soluble guanylyl cyclase.
Used to treat the symptoms, not the underlying cause, of angina.
Use of all nitrates is contra-indicated in patients suffering from aortic stenosis, cardiac tamponade, constrictive pericarditis, hypertrophic cardiomyopathy, hypotensive conditions, hypovolaemia, marked anaemia, mitral stenosis, raised intracranial pressure due to cerebral haemorrhage, raised intracranial pressure due to head trauma or toxic pulmonary oedema. Prescribers should be aware of tolerance developing in patients using long-acting or transdermal nitrates, and monitor blood nitrate levels to ensure low levels for 4-12 hours per day to maintain drug efficacy.
Common side effects for all nitrates include dizziness, postural hypotension, tachycardia and throbbing headache.
Glyceryl trinitrate (GTN): administered as sublingual tablets, sublingual aerosol spray, transdermal patches or intravenous infusion for treatment and prophylaxis of angina. GTN can also be used to teat unstable angina and congestive heart failure.
Isosorbide dinitrate: administered as immediate-release sublingual tablets, sublingual aerosol spray or intravenous infusion for prophylaxis or treatment of angina. Immediate-release and IV formulations can be used to treat left ventricular failure. Modified-release formulations are used for prophylaxis of angina. Isosorbide dinitrate is slower to act than GTN.
Isosorbide mononitrate: administered as sublingual tablets for prophylaxis of angina
RESOURCES
This 11-slide slide set created with PowerPoint describes the pharmacology of organic nitrates: mechanisms of action; routes of administration; common unwanted effects; mechanisms of tolerance and the interaction between organic nitrates and phosphodiesterase 5 inhibitors. This is an introduction to the topic of organic nitrates which would be appropriate for beginners. Contributed by Christopher Fowler, Umeå University, Sweden.
Learner level: Beginner
Other antianginal drugs
Stable angina usually results from atherosclerotic plaques in the coronary arteries. This restricts blood flow and oxygen supply to the heart. Stable angina is often precipitated by exertion and relieved by rest.
Stable angina medications include nitrates (the drugs of choice), calcium-channel blockers (CCBs), beta-blockers and potassium channel activators. These drug families have vasodilatory effects which reduce blood pressure. Arteriolar dilatation reduces peripheral vascular resistance and left ventricular pressure during systole resulting in improved cardiac output.
In patients with intolerance of, or contra-indication to, either nitrates, beta-blockers or CCBs, the drugs listed below can be used as alternative antianginal medications.
Potassium channel activators:
Nicorandil: a potassium-channel activator with a nitrate component, with both arterial and venous vasodilating properties. Approved for the prevention and long-term treatment of angina.
Drugs with ‘other’ mechanisms of action :
Ivabradine: Ivabradine inhibits the If ion current (a mixed Na+–K+ inward current activated by hyperpolarization and modulated by the autonomic nervous system) in the sinoatrial node (cardiac pacemaker) (PMID: 15301560). Heart rate is reduced without any negative effects on myocardial contractility or ventricular repolarization. Oral ivabradine is indicated for angina patients in normal sinus rhythm, and for those with mild to severe chronic heart failure. Contra-indicated in acute myocardial infarction or slow heart rate (<70bpm), immediately after cerebrovascular accident, patients dependent on pacemaker, second- and third-degree heart block, severe hypotension, sick-sinus syndrome, sino-atrial block, unstable angina or unstable or acute heart failure. Common side-effects include atrial fibrillation, blurred vision, bradycardia, dizziness, first-degree heart block, headache, phosphenes, ventricular extrasystoles and visual disturbances.
Ranolazine: Likely to act via cardiac sodium channels to modulate sodium ion permeability and cellular excitability. Indicated as an adjunctive therapy in the treatment of stable angina in patients inadequately controlled or intolerant of first-line antianginal therapies. Caution should be exercised in patients with low body weight, moderate to severe congestive heart failure, QT interval prolongation and in the elderly. Avoid use in patients with moderate and severe hepatic or renal impairment.Common side-effects include asthenia, constipation, dizziness, headache, nausea and vomiting.
Positive inotropic drugs: Catecholamines and PDE3 inhibitors
Positive inotropic agents increase myocardial contractility by increasing the level of calcium in the cytoplasm of the muscle cells or by increasing the sensitivity of the heart to calcium. Positive inotropes are indicated in acute conditions where there is low cardiac output (CO), such as cardiogenic shock following myocardial infarction, acute decompensated heart failure, low CO states after cardiac surgery, cardiogenic shock, septic shock, and cardiomyopathy. In contrast, negative inotropes weaken the force of contraction of the heart
There is limited evidence to suggest that one particular positive inotrope is better than another. The choice of which drug to prescribe will depend on factors such as the patient’s underlying disease state and the clinician’s preference.
Drugs that act as positive inotropes include the catecholamines (dobutamine and isoprenaline [synthetic catecholamines] and adrenaline and noradrenaline [endogenous catecholamines]) and phosphodiesterase type-3 (PDE3) inhibitors.
Cardiac effects of catecholamines are attributed to their action as agonists of alpha and beta adrenoceptors, in particular activation of the ß1-adrenoceptor increases heart contractility and heart rate. Dobutamine is predominantly a ß1 agonist, but its action at ß2 receptors causes vasodilation (and decreases afterload) which can be compensated by administering a vasopressor such as noradrenaline. Isoprenaline has a similar profile to dobutamine but tends to cause more tachycardia. Noradrenaline, acting mainly via α1-adrenoceptors, is primarily used as a vasopressor (increasing afterload to maintain mean arterial pressure) rather than an inotrope. It is often used with other inotropes, such as dobutamine, to maintain adequate perfusion (supra). As adrenaline has activity at all adrenoceptors other more specific inotropes are often preferred over adrenaline. The main use of adrenaline is as the bolus administered during resuscitation after cardiac arrest.
Inhibition of PDE3 increases intracellular calcium causing vasodilation and increased myocardial contractility and is the causative mechanism of action of the PDE3 inhibitors enoximone, milrinone and the non-selective phosphodiesterase inhibitor theophylline.
RESOURCES
This 24-slide slide set provides an introduction at new learner to intermediate level to some of the most common drugs that are used clinically to modulate the rate and force of contraction of the heart. The specific drug classes presented include: agonists and antagonists of beta-adrenoceptors, an antagonist of muscarinic acetylcholine receptors (i.e. atropine), cardiotonic agents including cardiac glycosides (i.e. digoxin) and miscellaneous other agents. This presentation does not include drugs used to treat cardiac arrhythmias (other than the use of digoxin in specific conditions). A condensed physiological recap of the cardiac action potential and excitation contraction coupling are also included to help the reader appreciate the molecular basis of drug action upon the heart. Slide set provided by John Peters, University of Dundee, UK.
This video is approximately 37 minutes long and discusses the role of inotropic drugs in treating congestive heart failure with the help of concept maps/diagrams and animations.This presentation starts with an overview of pathophysiological basis of treating congestive cardiac failure (CCF). Various drugs commonly used to treat CCF are listed to show how inotropic drugs fit in the larger picture. Then, a description of the mode of action, uses and side effects of inotropic agents and their role in CCF is given in greater detail. Finally, a summary is given. The prerequisite topics include, physiology and therapeutic application of sympathetic nervous system (with special reference to beta adrenoceptor stimulation and inhibition), renin-angiotensin aldosterone system, regulation of body water and ion balance, diuretics, and pathophysiology and clinical features of heart failure.
Intermediate level.
Kindly contributed by Dr. Nasir Ali Afsar (Sohail University, Karachi, Pakistan).
Positive inotropic drugs: Cardiac glycosides (digoxin)
Digoxin is a member of a class of drugs known as the cardiac glycosides that also includes digitoxin and ouabain. Cardiac glycosides occur naturally in plants of the genera Digitalis, such as foxgloves and Strophanthus. Only digoxin and very rarely digitoxin are used clinically. Such agents increase the force of contraction of the heart, a positive inotropic action which underlies their use in some cases of heart failure. They also have important effects upon electrical conduction in the heart, particularly the velocity at which the action potential is conducted through the atrioventricular (AV) node. Clinically, digoxin is used in severe heart failure, but usually as a third line agent when other treatments including ACE inhibitors, low dose β-blockers and aldosterone receptor antagonists, or angiotensin receptor blockers do not provide sufficient benefit. However, as explained below, digoxin may be introduced at an earlier stage if the patient has atrial flutter as a co-morbidity.
Relevant Chemistry
Structurally, the cardiac glycosides considered here (the cardenolides) consist of a steroid ring to which a lactone and sugar residues are attached (in the β-configuration) at the C17 (D ring) and C3 (A ring) positions, respectively. An unsaturated lactone ring is essential for the pharmacodynamic action of the cardiac glycosides, as is cis-fusion of the steroid A/B and C/D rings, trans-fusion of the B/C rings and the presence of a β-hydroxyl at C14. The sugar groups (which are variable in nature and number, e.g. one rhamnose in ouabain, or three digitoxose in digoxin and digitoxin) influence the potency and pharmacokinetics of individual compounds. The nature of the sugar also contributes to the modest selectivity of cardiac glycosides between isoforms at their primary molecular target, the Na+/K+-ATPase. The latter is classically regarded as a heterodimer of α (α1, α2, α3 and α4) and β (β1, β2 and β3) subunits in a 1:1 stoichiometry in association with regulatory subunits (phospholemman in the heart)
Mechanism of action
Cardiac glycosides bind to the catalytic α-subunit of the Na+/K+-ATPase (the ‘sodium pump’) inhibiting its action to transport Na+ out of, and K+ into, the cardiac muscle cell. Thus, therapeutic concentrations of digoxin bind to a proportion of the Na+/K+-ATPase pumps in cardiac muscle, reducing overall pumping activity. Excessive inhibition of the pump underlies many of the serious adverse effects of digoxin which limit its use (see below).
Binding of digoxin occurs at the extracellular side of the pump in competition with K+ explaining, at least partially, the clinically important phenomenon that a reduced concentration of K+ in the plasma (hypokalaemia) increases the action of digoxin which may precipitate serious toxicity. In addition, reduced plasma K+ may result in the phosphorylation of the Na+/K+-ATPase increasing its affinity for binding of digoxin and thus pump occupancy. Both α1β and α2β isoforms of the Na+/K+-ATPase have been implicated in the inotropic action of cardiac glycosides.
In cardiac muscle, which due to its high level of electrical activity is particularly reliant upon the Na+/K+-ATPase to maintain appropriate ion gradients across the plasma membrane, inhibition of the pump causes an elevation of the intracellular concentration of Na+ ([Na+]i). This is accompanied by a small reduction in the resting (diastolic) membrane potential of cardiac muscle cells because the pump is electrogenic (i.e. pumps 3 Na+ out: 2 K+ in for each transport cycle at the expense of one molecule of ATP hydrolysed to ADP and Pi). The subsequent reduction in the electrochemical gradient for Na+ entry secondarily reduces the expulsion of Ca2+ from the cytoplasm during diastole by the plasma membrane Na+/Ca2+ exchanger (NCX1) operating with the stoichiometry 3 Na+ in: 1 Ca2+ out during each transport cycle. This occurs because the operation of the Na+/K+-ATPase (primary active transport) is required to maintain the secondary active transport mediated by NCX1. The excess Ca2+ is sequestered into the lumen of the sarcoplasmic reticulum (SR) by the Ca2+-ATPase (SERCA2a) within the membrane of that organelle. Thus, additional free Ca2+ is available for release from the SR lumen during the plateau phase (phase 2) of the ventricular action potential by the process of calcium-induced calcium release (CICR). The latter process is elaborated upon in the textbox. The cytoplasmic Ca2+ transient that ensues and generates systole is thus elevated and via increased occupation of the cardiac isoform of troponin-C (TNNC1) by Ca2+ translates into an increase in cardiac contractility.

The effect of digoxin upon the electrical activity of the heart is complex and consists of a number of direct and indirect actions. Directly, inhibition of the Na+/K+-ATPase causes a small depolarization of cardiac muscle (see above) that predisposes to abnormal discharge of action potentials by cardiac muscle cells and consequently ectopic beats (see below). Acting directly upon the conduction system of the heart, digoxin slows conduction velocity through the atrioventricular (AV) node by prolonging the refractory period. The latter action is useful and underlies the use of the drug in patients with atrial fibrillation or flutter accompanied by a high ventricular rate, usually if combined with heart failure. Although digoxin does not correct atrial fibrillation or flutter, it suppresses aberrant impulses from conducting through the AV node to trigger ventricular arrhythmias. Conversely, excessive suppression of AV conduction leads to heart block, one of the characteristic features of digoxin toxicity. Indirectly, but again through inhibition of the Na+/K+-ATPase, digoxin stimulates the parasympathetic nerve supply to the heart by acting upon the central vagal nucleus (i.e. increases vagal tone) which reinforces its suppressant effect upon AV node conduction velocity and also decreases the automaticity of the sinoatrial (SA) node, thus reducing discharge rate. The latter action slows the heart rate in sinus rhythm.
Pharmacokinetics
Digoxin may be administered by mouth, or if a rapid action is required, by slow intravenous injection. When given by mouth, the bioavailability of digoxin is influenced by the formulation of the drug (tablets, or liquid). The drug has a high apparent volume of distribution, principally due to binding to the Na+/K+-ATPase of skeletal muscle, and coupled with an elimination half-life of approximately 36 hours (with normal renal function, see below) this necessitates a loading dose usually by mouth if a rapid action is required. The drug may be taken with, or without, food.
Digoxin is a polar molecule and the main route of elimination (approximately 70% of the drug unchanged) is via renal excretion involving both glomerular filtration and active tubular secretion. Thus, in significant renal impairment, the dose of digoxin must be reduced particularly in view of the very low therapeutic index of the drug. The reduction in dosage is proportional to the fall in glomerular filtration rate (GFR). Hepatic metabolism of digoxin contributes to elimination to a much lower extent than renal excretion.
Drug interactions
The margin of safety between the doses of digoxin that produce a therapeutic, or toxic, effect is very narrow (plasma concentrations of approximately 1 – 2.6 nmol/l are reported as the therapeutic window). Moreover, digoxin exhibits important pharmacodynamic and pharmacokinetic interactions with many drugs, including some used to treat heart disease. Some important examples are:
Pharmacodynamic – these are largely predictable from a basic understanding of cardiovascular pharmacology:
- with β-adrenoreceptor antagonists: AV node conduction velocity is facilitated by noradrenaline (and adrenaline) acting as agonists upon β1-adrenoceptors in nodal tissue. Blockade of this effect by β1-antagonists, in combination with the negative action of digoxin upon AV node conduction velocity, can lead to a high grade AV block. In addition, β-blockers exert a negative inotropic action that physiologically antagonises the inotropic action of digoxin.
- with Ca2+ channel blockers: Ca2+ channel blockers that exhibit a degree of selectivity for L-type Ca2+ channels in cardiac muscle (e.g. verapamil) can produce a negative inotropic action, negating the beneficial positive inotropic action of digoxin.
- with thiazide and loop diuretics: both classes cause loss of potassium from the body leading to hypokalaemia. As explained above, this potentiates the action of digoxin leading to potential toxicity.
Pharmacokinetic – these are less easily understood from a knowledge of mechanism and are probably best consulted in comprehensive textbooks of basic and clinical pharmacology and national and local formularies:
- with drugs that increase the absorption of digoxin from the gastrointestinal tract. Many antibiotics (e.g. erythromycin) may cause this by depleting intestinal flora that metabolise a fraction of the drug before it is absorbed across the mucosa.
- with drugs that alter the apparent volume of distribution, or renal clearance, of digoxin. Examples of particular relevance due to their cardiovascular/renal actions are verapamil, quinidine, amiodarone and spironolactone.
Adverse effects
The major adverse effects of digoxin include:
- bradycardia
- block of AV node conduction (see above): digoxin is contraindicated in patients with second degree heart block, or intermittent complete heart block
- triggering a variety of arrhythmias including ectopic beats (see above): delayed after depolarizations (DADs) may result from Ca2+ overload causing coupled beats (bigeminy)
Digoxin is contraindicated in patients who have, or are at risk of
- ventricular arrhythmias
- gastrointestinal disturbances such as anorexia, nausea, vomiting and diarrhoea
- neurological disturbances such as yellow vision (probably due to an action on the retina), fatigue, malaise and confusion.
Digoxin toxicity may be precipitated by electrolyte disturbances that include hypokalaemia (see above), hypomagnesaemia and hypercalcaemia. Treatment of digoxin toxicity includes withholding the drug, potassium supplementation to correct hypokalaemia and in severe cases the administration of digoxin specific antibody fragments.
Contributor: John Peters
RESOURCES
This 24-slide slide set provides an introduction at new learner to intermediate level to some of the most common drugs that are used clinically to modulate the rate and force of contraction of the heart. The specific drug classes presented include: agonists and antagonists of beta-adrenoceptors, an antagonist of muscarinic acetylcholine receptors (i.e. atropine), cardiotonic agents including cardiac glycosides (i.e. digoxin) and miscellaneous other agents. This presentation does not include drugs used to treat cardiac arrhythmias (other than the use of digoxin in specific conditions). A condensed physiological recap of the cardiac action potential and excitation contraction coupling are also included to help the reader appreciate the molecular basis of drug action upon the heart. Slide set provided by John Peters, University of Dundee, UK.
This video is approximately 37 minutes long and discusses the role of inotropic drugs in treating congestive heart failure with the help of concept maps/diagrams and animations.This presentation starts with an overview of pathophysiological basis of treating congestive cardiac failure (CCF). Various drugs commonly used to treat CCF are listed to show how inotropic drugs fit in the larger picture. Then, a description of the mode of action, uses and side effects of inotropic agents and their role in CCF is given in greater detail. Finally, a summary is given. The prerequisite topics include, physiology and therapeutic application of sympathetic nervous system (with special reference to beta adrenoceptor stimulation and inhibition), renin-angiotensin aldosterone system, regulation of body water and ion balance, diuretics, and pathophysiology and clinical features of heart failure.
Intermediate level.
Contributor by: Dr. Nasir Ali Afsar (Sohail University, Karachi, Pakistan).
This 7 minute animation reviews the important organs, hormones, enzymes and mechanisms by which the RAAS system controls blood pressure.