
Ion channels
Ion channels are pore-forming protein complexes that facilitate the flow of ions across the hydrophobic core of cell membranes. They are present in the plasma membrane and membranes of intracellular organelles of all cells, performing essential physiological functions including establishing and shaping the electrical signals which underlie muscle contraction/relaxation and neuronal signal transmission, neurotransmitter release, cognition, hormone secretion, sensory transduction and maintaining electrolyte balance and blood pressure. They are usually classified by gating i.e. the stimulus that 'opens' the channel, be it chemical or mechanical stimuli.
Most Na+, K+, Ca2+ and some Cl- channels are gated by voltage, whereas others (such as some K+ and Cl- channels, TRP channels, ryanodine receptors and IP3 receptors) are relatively voltage-insensitive and are gated by second messengers and other intracellular and/or extracellular mediators.
Many ion channels (e.g. K+, Na+, Ca2+, HCN and TRP channels) share several structural similarities which suggests that they have evolved from a common ancestor. This group of ion channels can be classified together as the ‘voltage-gated-like (VGL) ion channel chanome’ (Figure 1).

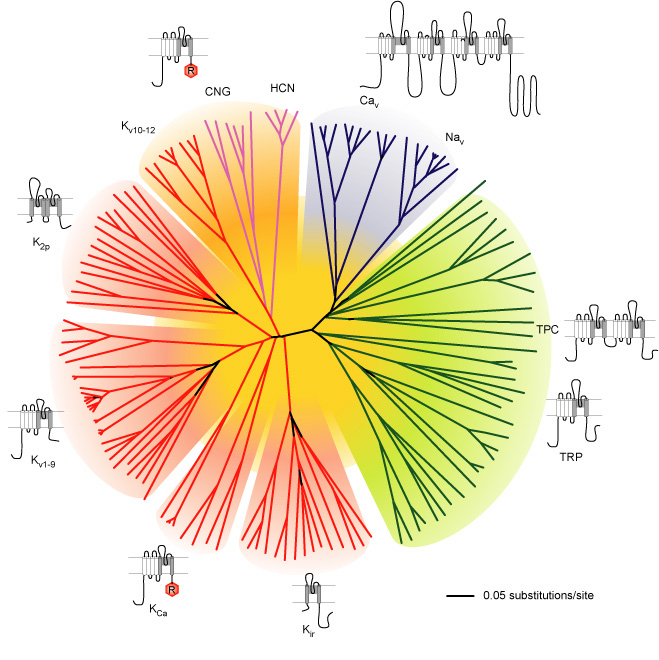
Figure 1. Representation of the amino acid sequence relations of the minimal pore regions of the voltage-gated ion channel superfamily. This global view of the 143 members of the structurally related ion channel genes highlights seven groups of ion channel families and their membrane topologies. Four-domain channels (CaV and NaV) are shown as blue branches, potassium selective channels are shown as red branches, cyclic nucleotide–gated channels are shown as magenta branches, and transient receptor potential (TRP) and related channels are shown as green branches. Background colors separate the ion channel proteins into related groups: light blue, CaV and NaV ; light green, TRP channels; light red, potassium channels, except KV10–12, which have a cyclic nucleotide–binding domain and are more closely related to CNG and HCN channels; light orange, KV10–12 channels and cyclic nucleotide–modulated CNG and HCN channels (from Yu and Catterall, 2004)
Other ion channels, such as Cl channels, aquaporins and connexins, have evolved separately and possess completely different structural properties to the VGL channels.
Defects in ion channel function cause a wide range of disorders termed 'channelopathies' which include conditions resulting from mutations in ion channels (e.g. cystic fibrosis, long QT syndrome, short QT syndrome- inherited ion channel diseases are reviewed by Lieve and Wilde (2015)) and acquired diseases caused by autoimmune attack on ion channels (e.g. myasthenia gravis and possibly multiple sclerosis- autoimmunity and channelopathy are reviewed by RamaKrishnan and Sankaranarayanan (2016)).
Ion channel modulators are an extremely successful drug class, second only to drugs targeting G protein-coupled receptors, with amlodipine, zolpidem, alprazolam, the sulfonylureas, repaglinide and nateglinide amassing huge returns for their developers. Technical advances in high-throughput screening methodology and high resolution crystal structures of ion channels should enable development of the ion channel drugs of the future. Potential new ion channel drug targets are discussed in Bagal et al. (2013).
